The history of self-absorption correction¶
(written for VIPER and XANES dactyloscope in April 2012)
The early papers by [Goulon], [Tan], [Tröger] were limited only to the EXAFS case. The correction functions there had discontinuity at the edge and thus were not applicable to XANES. Moreover, those works provided corrections only for infinitely thick samples with an exception of [Tan et al. 1989] where also thin samples were considered but only as pure materials (e.g. single element foils).
The first self-absorption correction for the whole absorption spectra (also including XANES) was proposed with two different strategies by [Eisebitt] and [Iida]. [Eisebitt] estimated the two unknowns \(μ_{tot}\) and \(μ_X\) (see the notations below) from two independent fluorescence measurements with different positioning of the sample relative to the primary and fluorescence beams. An obvious disadvantage of this method is that it is solely applicable to polarization-independent structures (amorphous or of cubic symmetry). On the other hand, it does not require any theoretical tabulation, which is the case in the method of [Iida], who proposed the background part \(μ_{back} = μ_{tot} - μ_X\), to be taken as tabulated. The advantage of their approach is its applicability to any sample with only one measurement. Moreover, this method is applicable to samples of general thickness, not only to thick samples as required by the method of [Eisebitt]. It is the method of [Iida] which is implemented, with some variations, in ParSeq-XAS. The method was re-invented (i.e. published without citing [Iida]) by [Pompa], [Haskel] and [Carboni]. These three works, however, were simplified down to infinitely thick limit.
The correction was extended somewhat by considering a variable escape angle in order to account for the finite (not infinitely small) detector area: only in the synchrotron orbit plane, in EXAFS [Brewe] and also out of plane: in EXAFS [Pfalzer] and XANES [Carboni]. All three works operated in the thick limit. In my opinion, detector elements are always small enough in the sense that the self-absorption effect can be considered as uniform over each single element and therefore the correction can be done only for one direction toward the pixel center.
An interesting approach to correcting the self-absorption effect was proposed by [Booth] who considered another small parameter, not the usual exp(-μd), which allowed simplifying the formulas also beyond the thick limit but the treatment was limited to EXAFS.
Another re-invention of the Iida and Noma method with calling it “new” was presented by [Ablett]. The merit of that work was implementing the method without the restriction of the thick limit and providing many application examples and literature references.
Goulon J, Goulon-Ginet C, Cortes R and Dubois J M (1982) J. Physique 43, 539.
Tan Z, Budnick J I and Heald S M (1989) Rev. Sci. Instrum. 60, 1021.
Tröger L, Arvanitis D, Baberschke K, Michaelis H, Grimm U and Zschech E, (1992) Phys. Rev. B 46, 3283.
Eisebitt S, Böske T, Rubensson J-E and Eberhardt W (1993) Phys. Rev. B 47, 14103.
Pompa M, Flank A-M, Delaunay R, Bianconi A and Lagarde P (1995) Physica B 208&209, 143.
Haskel D (1999) Computer program FLUO: Correcting XANES for self absorption in fluorescence data, www3.aps.anl.gov/~haskel/fluo.html
Carboni R, Giovannini S, Antonioli G and Boscherini F (2005) Physica Scripta T115, 986.
Brewe D L, Pease D M and Budnick J I (1994) Phys. Rev. B 50, 9025.
Pfalzer P, Urbach J-P, Klemm M, Horn S, denBoer M L, Frenkel A I and Kirkland J P (1999) Phys. Rev. B 60, 9335.
Booth C H and Bridges F (2005) Physica Scripta T115, 202.
Description of self-absorption correction¶
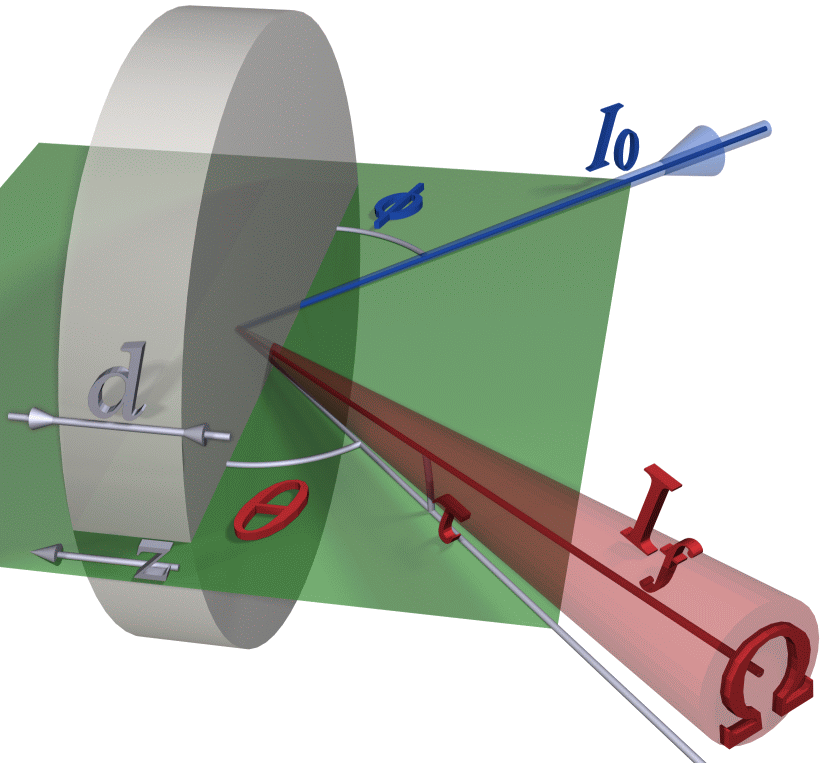
The derivation of the fluorescence intensity can be found, with different notations, in almost all the papers cited above. Here it is repeated because ParSeq-XAS adds some extra factors. The standard expression for the fluorescence intensity originated from a layer \(dz\) at the depth \(z\) is given by the trivial sequence of propagation and absorption (with neglected scattering):
where \(μ_T\) is the total linear absorption coefficient at the primary x-ray energy \(E\) or the fluorescence energy \(E_f\), \(μ_X\) is the contribution from the edge of interest, \(\epsilon_f\) is the fluorescence quantum yield – the probability to create a fluorescence photon from an absorbed photon. After integration over \(z\) from 0 to \(d\):
where the constant \(C\) includes all the energy independent factors and is treated as unknown because the actual solid angle is usually unknown and also because it implicitly includes the detector efficiency. The total absorption coefficient is decomposed as \(\mu_T=\mu_X+\mu_b\), where the background absorption coefficient \(μ_b\) is due to all other atoms and other edges of the element of interest. The constant \(C\) is found by equalizing all μ’s at a selected normalization energy to the tabulated ones. Now the last equation can be solved for \(μ_X\) at every energy point \(E\), which is the final goal of the self-absorption correction. When the sample is thick (\(d\to\infty\)), the exponent factors vanish. This “thick limit” approximation allows finding \(μ_X\) by a simple inversion of the equation and is optional in ParSeq-XAS.
Implementation in ParSeq-XAS¶
Some options offered by ParSeq-XAS are not quite standard (extended):
The additional term \(\cos\tau\) in is not quite standard; one can also find it in [Carboni] and [Ablett].
\(μ_b\) is usually taken as energy independent. In ParSeq-XAS it is energy dependent.
One can select among four different tabulations of absorption coefficients (actually, scattering factors \(f''\)) in ParSeq-XAS.
In order to use the equation for \(μ_X\), it is prerequisite to know the sample stoichiometry, i.e. the molar weighting factors \(x_i\) for each atom type \(i\) in the sample. The linear absorption coefficient is proportional to the atomic absorption cross section \(σ_a\): \(μ_X\propto x_Xσ_{aX}\) and \(μ_T\propto\sum_ix_iσ_{ai}\). The atomic cross sections, in turn, are calculated from the tabulated scattering factors \(f''\): \(σ_a=2r_0chN_Af''/E\).
Because all the tabulations do not contain the partial contributions of each absorption edge of an element but only the combined result of all atomic shells, an isolation of \(μ_X\) and the pre-edge background is required. In ParSeq-XAS this is done by extrapolating the pre-edge region by the Victoreen polynomial. The polynomial coefficients are found over only two pre-edge points, as the tabulations are usually sparse. The edge jump is the difference between the first post-edge value and the extrapolated background.
